
As mitocôndrias são a força motriz das células. Eles usam oxigênio para oxidar nutrientes por meio de quatro complexos de cadeia respiratória que manipulam elétrons e prótons para gerar energia na forma de ATP. Portanto, qualquer condição que afete a produção de energia mitocondrial tem o potencial de reduzir a energia disponível para os processos fisiológicos celulares, o que pode induzir a apoptose.
Em condições normais (lado esquerdo da figura), tanto ácidos graxos, quanto glicose que chegam da dieta ou são produzidos, alimentam o ciclo de Krebs para geração de energia (ATP), NADH e FADH2. A oxidação do NADH fornece elétrons para o primeiro complexo ligado à membrana na cadeia respiratória mitocondrial, enquanto a oxidação do FADH2 fornece elétrons para o segundo complexo.
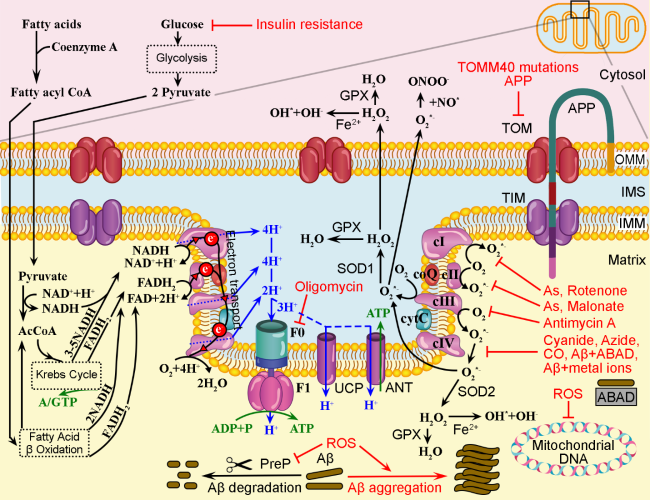
Os elétrons são transportados para a coenzima Q (coQ), depois para o complexo III (citocromo C) e complexo V, onde são usados para combinar oxigênio (O2) e prótons, gerando água (H2O). Quando os elétrons são transferidos para os complexos I, III e IV, é criado um gradiente de prótons, que depois retornam à matriz passando pela ATP sintase formada por duas subunidades denominadas F0 e F1, que gera o ATP que será utilizado para todas as inúmeras reações bioquímicas imagináveis. Em condições desfavoráveis (à direita), a resistência à insulina pode diminuir o transporte de glicose para o citoplasma dos neurônios, o que afetará a cadeia de transporte de elétrons e a produção de ATP.
Disfunção mitocondrial e doença de Alzheimer
Existem condições mitocondriais humanas que estão ligadas à doença de Alzheimer. Mutações em genes mitocondriais chave que afetam o transporte de proteínas ou nutrientes reduziriam a quantidade de combustível para os ciclos de Krebs e de β-oxidação de ácidos graxos, diminuindo assim a quantidade de ATP gerada pelas mitocôndrias. Uma diminuição crônica de ATP nos neurônios levará à produção de radicais livres de oxigênio (ROS) e nitrogênio (RNS). Assim mais proteína beta amilóide (Aβ) entra na célula na tentativa de guiar mitocôndrias ineficientes para a mitofagia.
Por exemplo, polimorfismos no gene TOMM40 estão associados a um início precoce e aumento do risco de desenvolver DA de início tardio. Os prótons podem vazar para a matriz através de outras proteínas, como a proteína desacopladora (UCP) e as translocases de nucleotídeos de adenina do trocador de ATP (ANT). Em situações fisiológicas, baixas quantidades de Aβ podem entrar nas mitocôndrias, mas são degradadas pela protease de pré-sequência (PreP). Já as mutações TOMM40 e a presença de proteína beta amilóide (APP) nos supercomplexos TOM/TIM podem impedir o transporte de proteínas para a matriz mitocondrial.
Outra condição mitocondrial ligada à DA é o desacoplamento da geração de elétrons e prótons por altos níveis de íons metálicos, toxinas e produtos químicos. As toxinas ambientais mais comuns geradas por atividades industriais e agrícolas são pesticidas, fumaça, micotoxinas, bifenilos policlorados (PCBs) e arsênico, além de poluentes ambientais, incluindo metais pesados. As toxinas podem inibir os complexos da cadeia respiratória mitocondrial e os íons metálicos livres podem aumentar a produção de EROs, que têm sido diretamente ligadas à toxicidade neuronal, síntese de Aβ e aumento do risco de desenvolver DA.
Por exemplo, o inseticida rotenona pode inibir o transporte adequado de elétrons do complexo I (cI), que se combinará com O2 para formar ânions radicais superóxido (O2*-). Os compostos naturais e industriais malonatos inibem o complexo II (cII) e induzem a formação de O2*-. O arsênio (As) pode inibir tanto cI quanto cII. A toxina Antimicina A produzida pela bactéria Streptomyces pode inibir cIII. Poluentes industriais como cianeto, azida, CO, bem como Aβ endógeno ligado a proteínas da matriz como Aβ álcool desidrogenase de ligação (ABAD) ou íons metálicos podem inibir cIV. A oligomicina A, também produzida pela bactéria Streptomyces, é um inibidor de F0, inibindo assim a produção de ATP. Os ânions do radical superóxido podem ser catalisados pela superóxido dismutase 1 (SOD1) no IMS, ou SOD2 na matriz, em O2 e peróxido de hidrogênio (H2O2).
Fisiologicamente ROS e RNS são produzidos em níveis baixos e neutralizados por antioxidantes não enzimáticos e enzimáticos (por exemplo, glutationa, flavonóides, superóxido dismutase e catalases). Mas quando os níveis de ROS aumentam demais, os mecanismos antioxidantes podem ficar sobrecarregados. Durante o envelhecimento, as mitocôndrias e outras organelas liberam cada vez mais ROS, que podem danificar o DNA e o RNA, além de inibir o PreP, o que estimulará a agregação de Aβ dentro da matriz mitocondrial. O aumento nos níveis de ROS também decorre da ativação microglial relacionada à Aβ, inflamação e ligação de metais redox. Por outro lado, tanto a atividade de enzimas antioxidantes quanto a ingestão de antioxidantes não enzimáticos diminuem com o envelhecimento, o que exacerba o aumento geral de ROS e RNS no corpo e no cérebro (Decourt et al., 2022).
