A perimenopausa não deve ser tratada apenas como que da hormonal. O artigo Personalized nutrition and precision medicine in perimenopausal women: A minireview of genetic polymorphisms COMT, FUT2, and MTHFR explica isso (Andrade et al., 2025). Genes isolados não explicam sozinhos sintomas clínicos. Nenhum gene atua isoladamente. Genes funcionam dentro de vias metabólicas integradas. O efeito clínico não vem de uma única variação e sim de vias alteradas.
Genes interagem o tempo todo e na perimenopausa isso não é diferente. Pequenas variações mudam o metabolismo do estrogênio, a resposta ao estresse, a absorção de B12, metilação e níveis de homocisteína, microbiota intestinal, regulação do cortisol. TRH não resolve tudo, é a precisão que muda o tratamento.
Por exemplo, COMT não afeta apenas dopamina. Ele participa da via de metilação e do metabolismo de catecolaminas e estrogênios. O impacto depende da disponibilidade de cofatores, da atividade de outras enzimas e do contexto fisiológico. COMT lenta gera mais sensibilidade ao estrogênio e ao estresse, com mais irritabilidade, ansiedade e insônia. Em um contexto de estrogênio instável, variantes em COMT só se manifestam clinicamente se a via de metilação estiver sobrecarregada ou deficiente. Na prática, olhar também a via da metilação e trabalhar junto. Adequar terapia, com doses menores de hormônios, via transdérmica e suporte à metilação.
O FUT2 não é um gene intestinal isolado. Ele influencia a composição da microbiota, a absorção de vitamina B12 e, indiretamente, vias imunometabólicas. O efeito clínico depende da interação gene–microbiota–nutrientes. Na menopausa, alterações intestinais e inflamatórias tornam essa via crítica para energia, cognição e resposta imune. Na prática, o paciente pode precisar de probióticos selecionados, prebióticos e B12 na forma ativa.
O MTHFR não determina sozinho a capacidade de metilação. Ele atua dentro da via do folato, integrada ao ciclo da metionina, ao metabolismo da homocisteína e à regulação epigenética. As variantes patogênicas de rs1801133 e 131 afetam a conversão do folato, aumentam a homocisteína, o risco cardiovascular. Na prática, pode ser necessário suplementar metilfolato, P5P, B12 ativa e monitorar risco cardiovascular. Lembre de avaliar também os outros genes da via do folato.
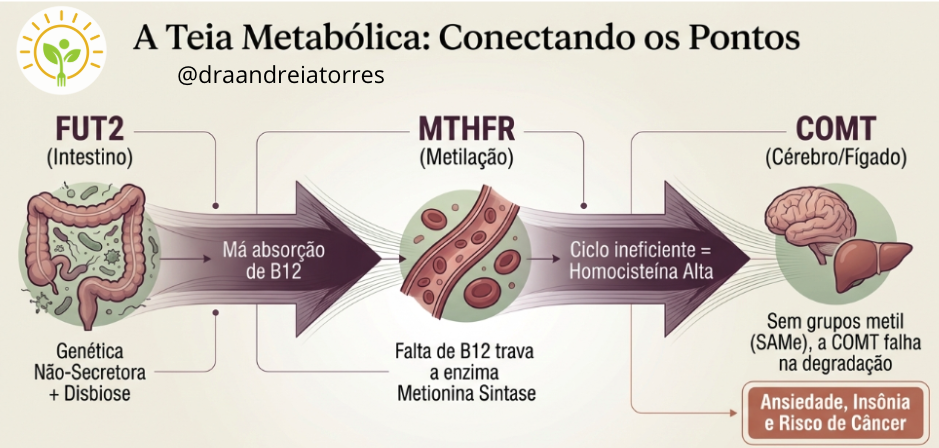
COMT, FUT2 e MTHFR funcionam de forma integrada e é essa interação influencia o bem estar da mulher, a metabolização de hormônio, os níveis de estresse e de inflamação. Sintomas como fadiga, ansiedade, névoa mental, ganho de peso e piora do sono emergem da convergência dessas vias, não de um gene específico. A vida em qualquer fase, incluindo perimenopausa e menopausa, exige leitura integrada de vias hormonais inflamatórias e de metilação. Genômica clínica não é lista de SNPs. É visualização de sistemas.
Curso: Genômica Visual. https://bit.ly/genomica-visual