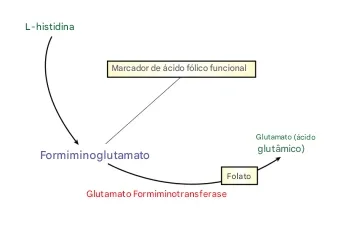
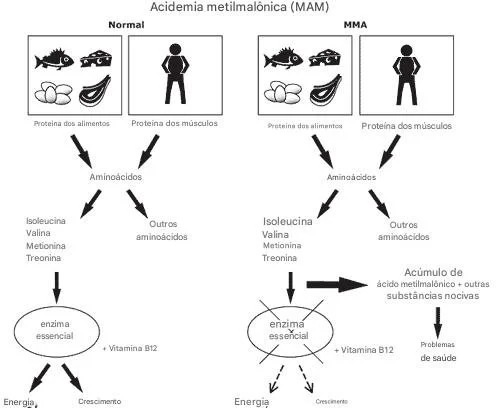
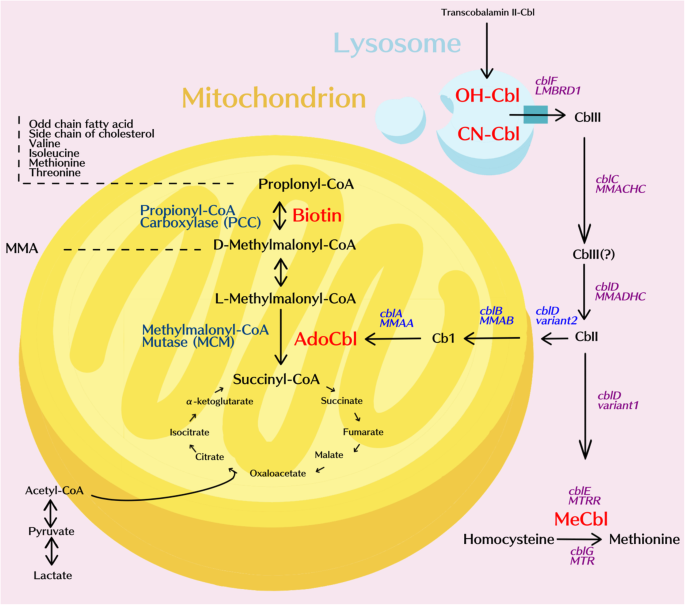
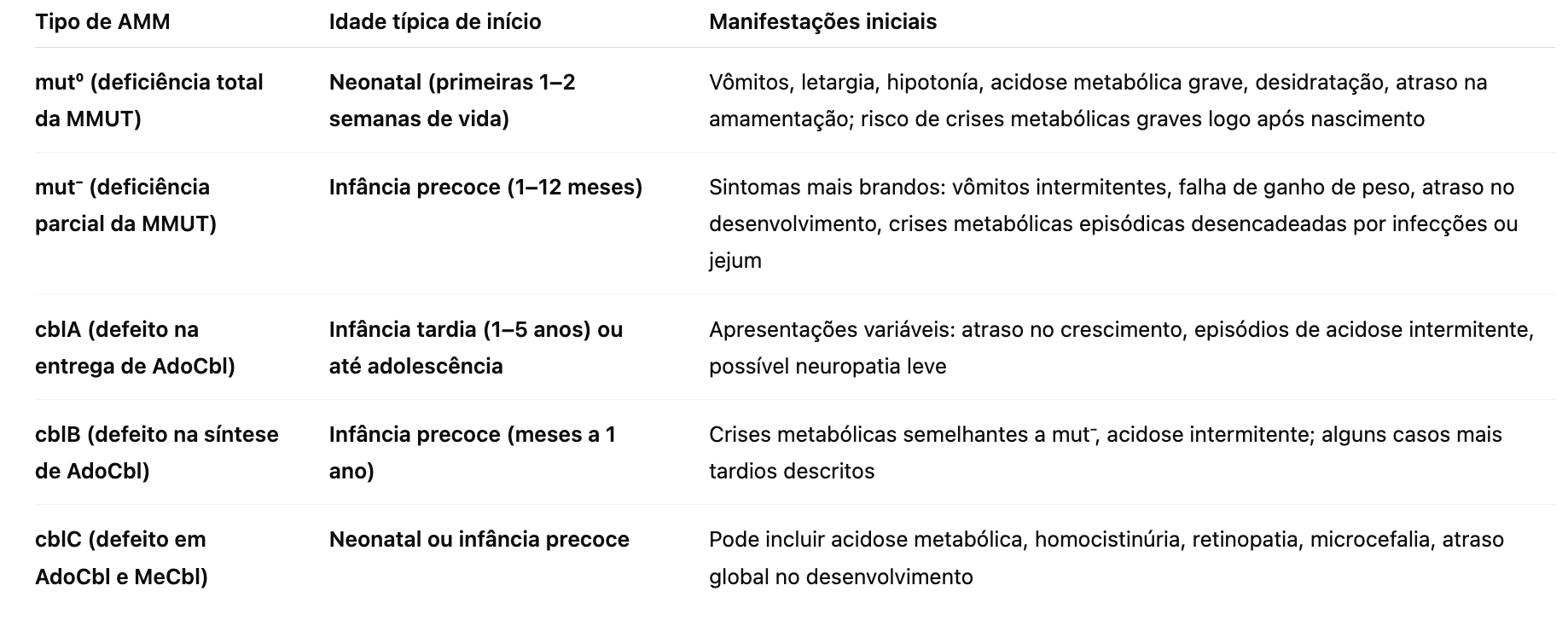
O artigo Metabolomics Approach Reveals Integrated Metabolic Network Associated with Serotonin Deficiency (Weng et al., 2015) utilizou uma abordagem metabolômica para identificar biomarcadores e vias metabólicas alteradas associadas à deficiência de serotonina induzida em camundongos por inibidores farmacológicos e alteração genética.
Identificação de biomarcadores:
pCPA: 21 biomarcadores alterados.
Tph2-/-: 17 biomarcadores alterados.
Alterações metabólicas observadas:
Aminoácidos: Alterações nos níveis de aminoácidos essenciais e não essenciais.
Metabolismo energético: Diminuição nos níveis de intermediários do ciclo de Krebs e ATP.
Purinas: Redução nos níveis de nucleotídeos purínicos.
Lipídios: Alterações nos perfis de ácidos graxos e lipídios complexos.
Microbiota intestinal: Indicações de alterações no metabolismo relacionadas à microbiota.
Estresse oxidativo:
Aumento nos níveis de espécies reativas de oxigênio (ROS) e produtos de peroxidação lipídica, sugerindo um estado pró-oxidante.
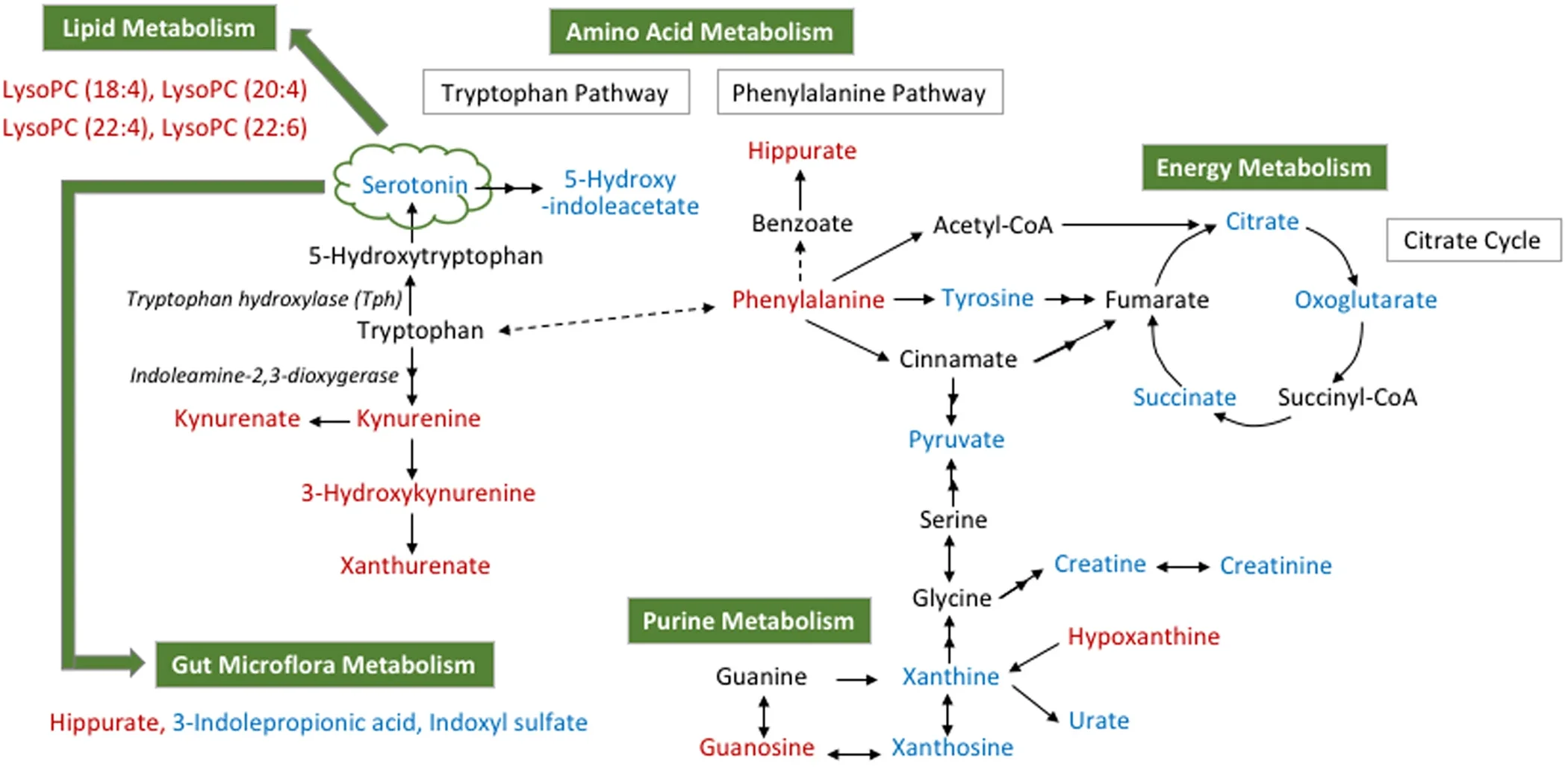
Vermelho: metabólitos que aumentam na deficiência de serotonina (ou marcadores alterados).
Azul: metabólitos reduzidos.
Preto: conexões enzimáticas ou caminhos principais.
Setas verdes: indica fluxos metabólicos que ligam serotonina a outras vias (lipídios, microbiota).
Integração das Vias Metabólicas
O estudo propõe uma rede metabólica integrada associada à deficiência de serotonina, destacando:
Interconexões entre vias: Como o metabolismo de aminoácidos influencia o metabolismo energético e lipídico.
Impacto no ciclo de Krebs: Diminuição na produção de energia celular.
Alterações na sinalização celular: Possíveis implicações na função neuronal e comunicação celular.
Implicações Clínicas
As alterações metabólicas observadas podem contribuir para condições como depressão e esquizofrenia. A deficiência de serotonina pode estar associada a processos patológicos em doenças como Alzheimer e Parkinson. A identificação de novos biomarcadores pode auxiliar no desenvolvimento de terapias direcionadas (Weng et al., 2015).