O artigo publicado na Translational Psychiatry investiga como a interação entre ritmos biológicos e metabolismo celular pode contribuir para a fisiopatologia do transtorno do espectro do autismo (TEA).
O estudo propõe um modelo integrador baseado em três eixos:
Desregulação dos ritmos circadianos
Alterações em vias de sinalização neuronal (WNT/β-catenina)
Reprogramação metabólica cerebral (glicólise aeróbica)
Esses sistemas interagem e amplificam alterações no neurodesenvolvimento.
Ritmo circadiano e cérebro
Os ritmos circadianos regulam:
expressão gênica
metabolismo energético
desenvolvimento neural
comportamento
Alterações de vias e genes do relógio podem desregular o ritmo circadiano:
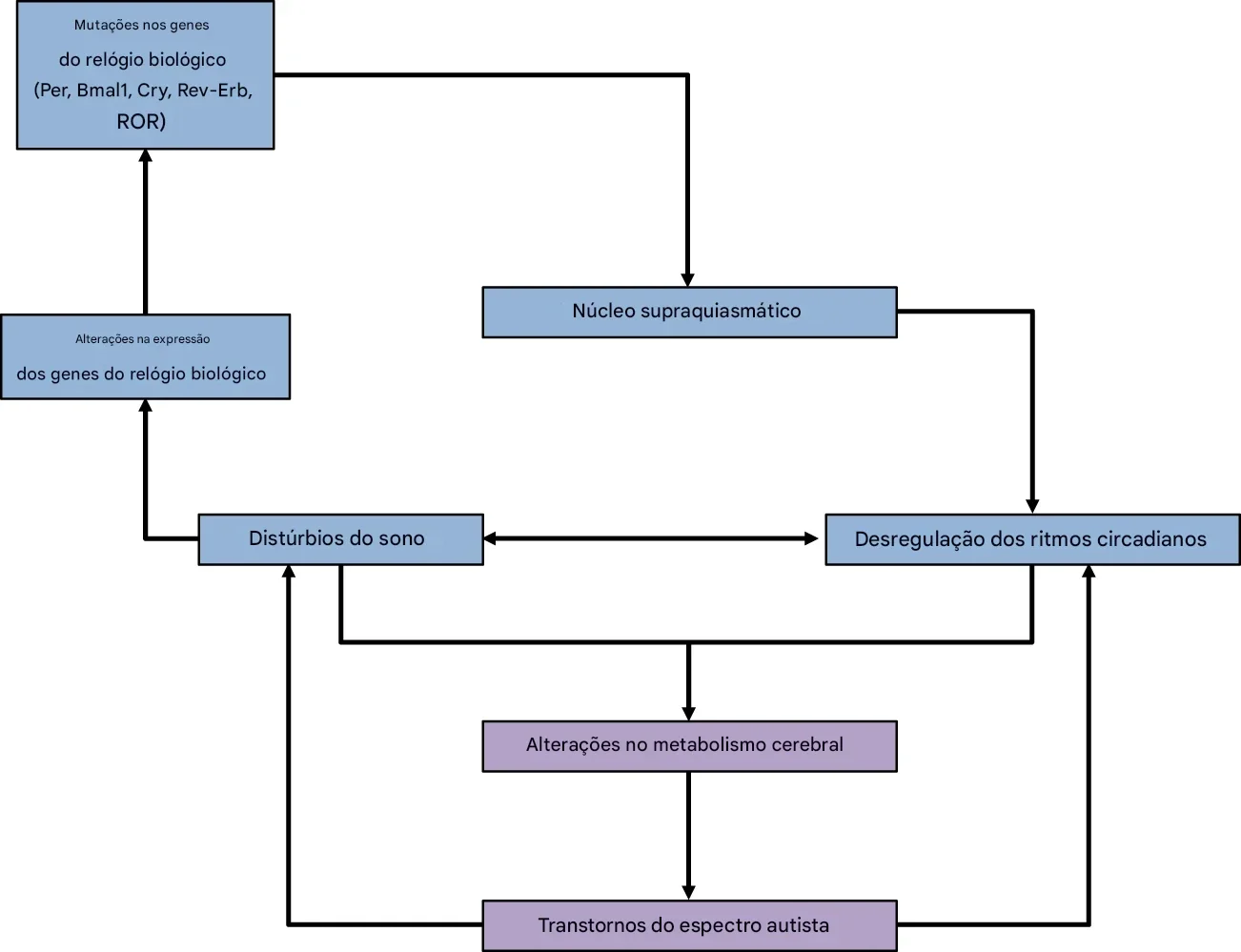
No TEA, há evidência de:
distúrbios do sono frequentes
alteração no “relógio biológico”
disfunção na sincronização entre ambiente e fisiologia
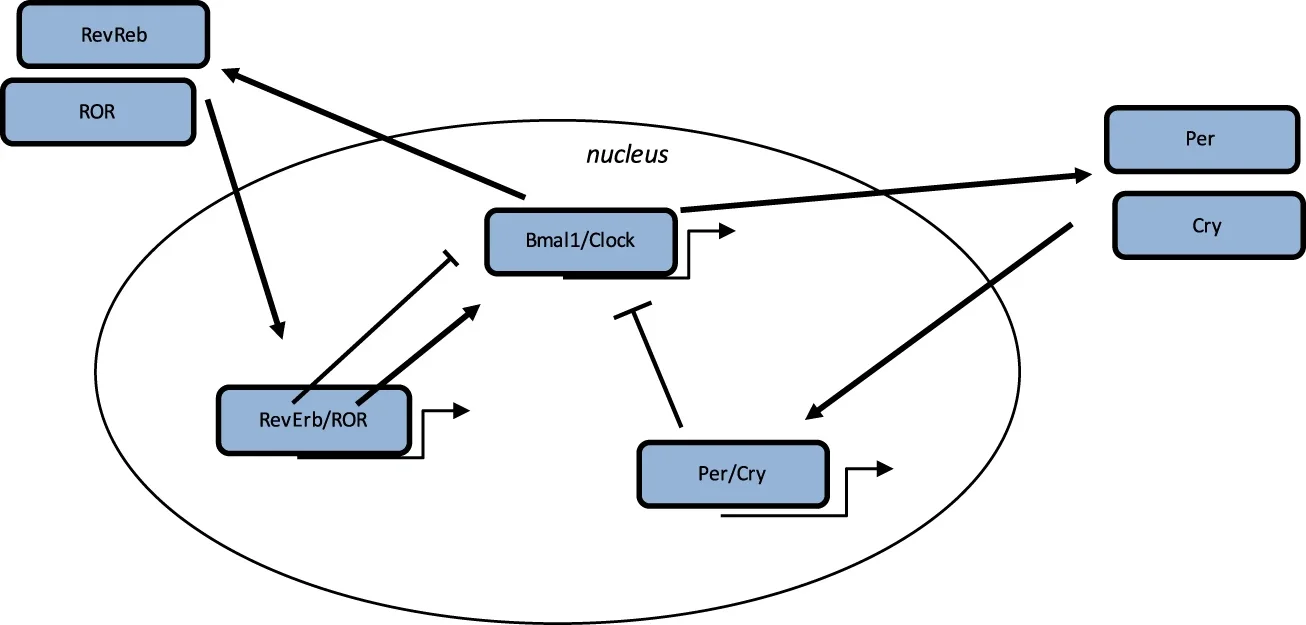
O processo do relógio biológico é um ciclo estimulatório, envolvendo o heterodímero Bmal1/Clock, que ativa a transcrição dos genes Per e Cry, e o ciclo de retroalimentação inibitória com o heterodímero Per/Cry, que se transloca para o núcleo e reprime a transcrição dos genes Clock e Bmal1. Um ciclo adicional envolve os fatores ROR e Rev-Erbs, com retroalimentação positiva por RORs e retroalimentação negativa por Rev-Erbs (Vallée et al., 2020)
Essa desorganização afeta diretamente circuitos cerebrais em formação, como a WNT/β-catenina, que é fundamental para o neurodesenvolvimento. Estas vias não funcionam de forma constante, são moduladas pelo ritmo circadiano.
Via canônica WNT/β-catenina
Via canônica é a forma clássica, mais bem caracterizada e funcionalmente dominante de uma via de sinalização celular. A via WNT/β-catenina é essencial para:
proliferação neuronal
diferenciação celular
formação de conexões sinápticas
Regulação da expressão de genes
Esta via é regulada por um conjunto de proteínas, como WNT e β-catenina. Quando não há sinal da WNT, a via fica inativa e a β-catenina livre no citoplasma liga-se a um complexo de destruição, formado por:
APC
AXIN
GSK-3β
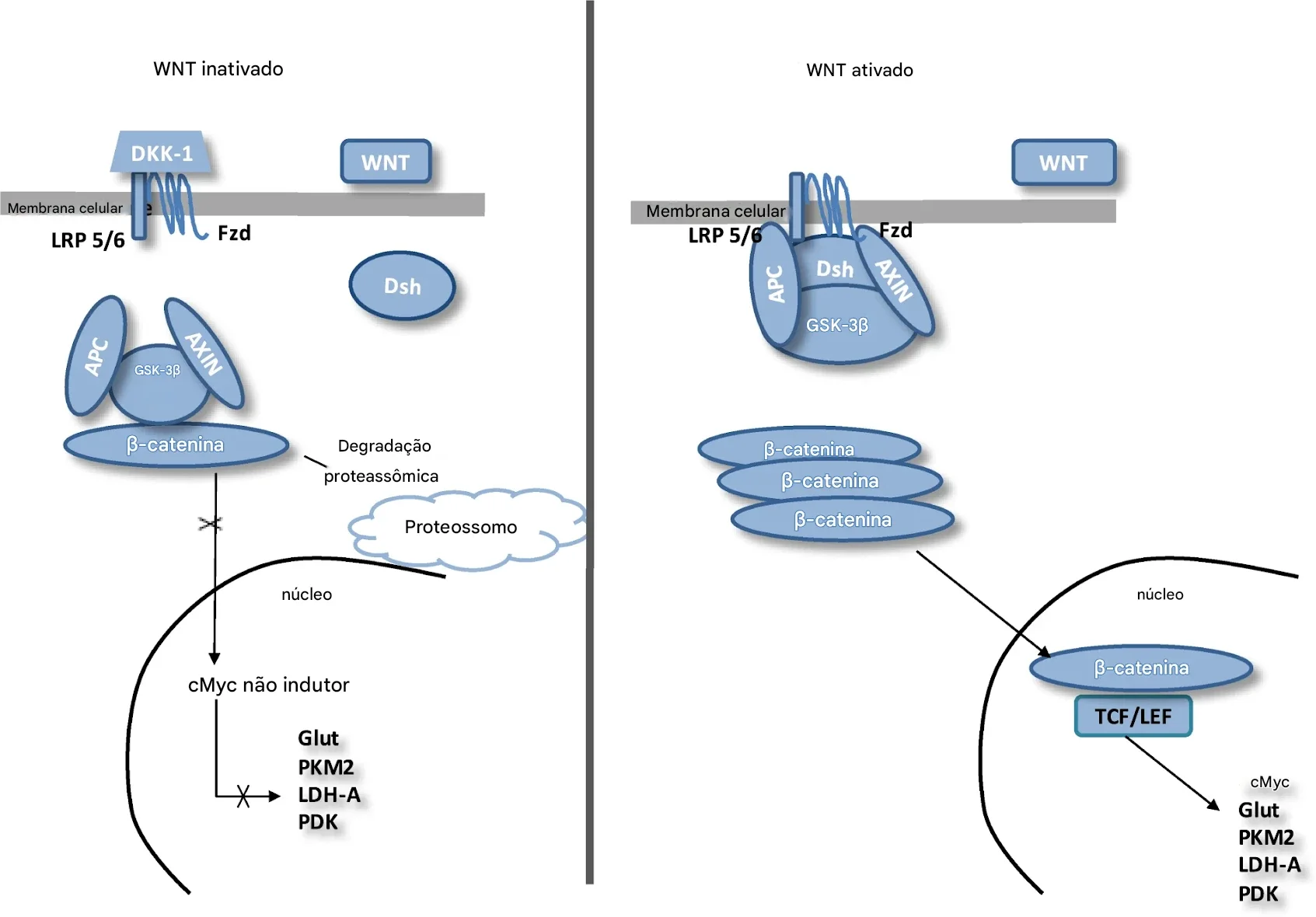
A β-catenina é fosforilada pela GSK-3β e degradada no proteassoma. Consequentemente, o nível citoplasmático de β-catenina é mantido baixo na ausência de ligantes WNT. Se a β-catenina não se acumula no núcleo, não estimula os genes-alvo. A consequência é o silenciamento da transcrição dos genes e redução da proliferação celular. Isso significa que alterações nessa via podem prejudicar o neurodesenvolvimento pela redução na proliferação de neurônios, diferenciação neuronal, formação de sinapses, organização de circuitos renais.
Quando WNT é ativado, a proteína DSH é estimulada e fosforilado pelo receptor FZD (frizzled). O DSH fosforilado, por sua vez, ativa a AXIN, que se dissocia do complexo de destruição da β-catenina. Assim, a β-catenina escapa da fosforilação e se acumula no citoplasma. A β-catenina citosólica acumulada se move para o núcleo, onde interage com os fatores de transcrição nucleares TCF/LEF e estimula a transcrição de genes-alvo.
O neurodesenvolvimento exige um padrão dinâmico. A via não pode ficar constantemente ligada ou desligada. No TEA, também pode ocorrer hiperativação dessa via. Se isso acontece, as ocorrências serão:
alterações cognitivas
desenvolvimento atípico do cérebro
A desregulação circadiana pode amplificar essa ativação, criando um ciclo patológico. A desorganização do padrão temporal, com muita ou pouca atividade podem gerar problemas:
ativação precoce excessiva → proliferação desorganizada
manutenção prolongada de sinal proliferativo → atraso de diferenciação
redução tardia inadequada → falha de maturação sináptica
A via WNT/β-catenina não regula apenas o destino celular, mas também reprograma o modo de produção de energia celular.
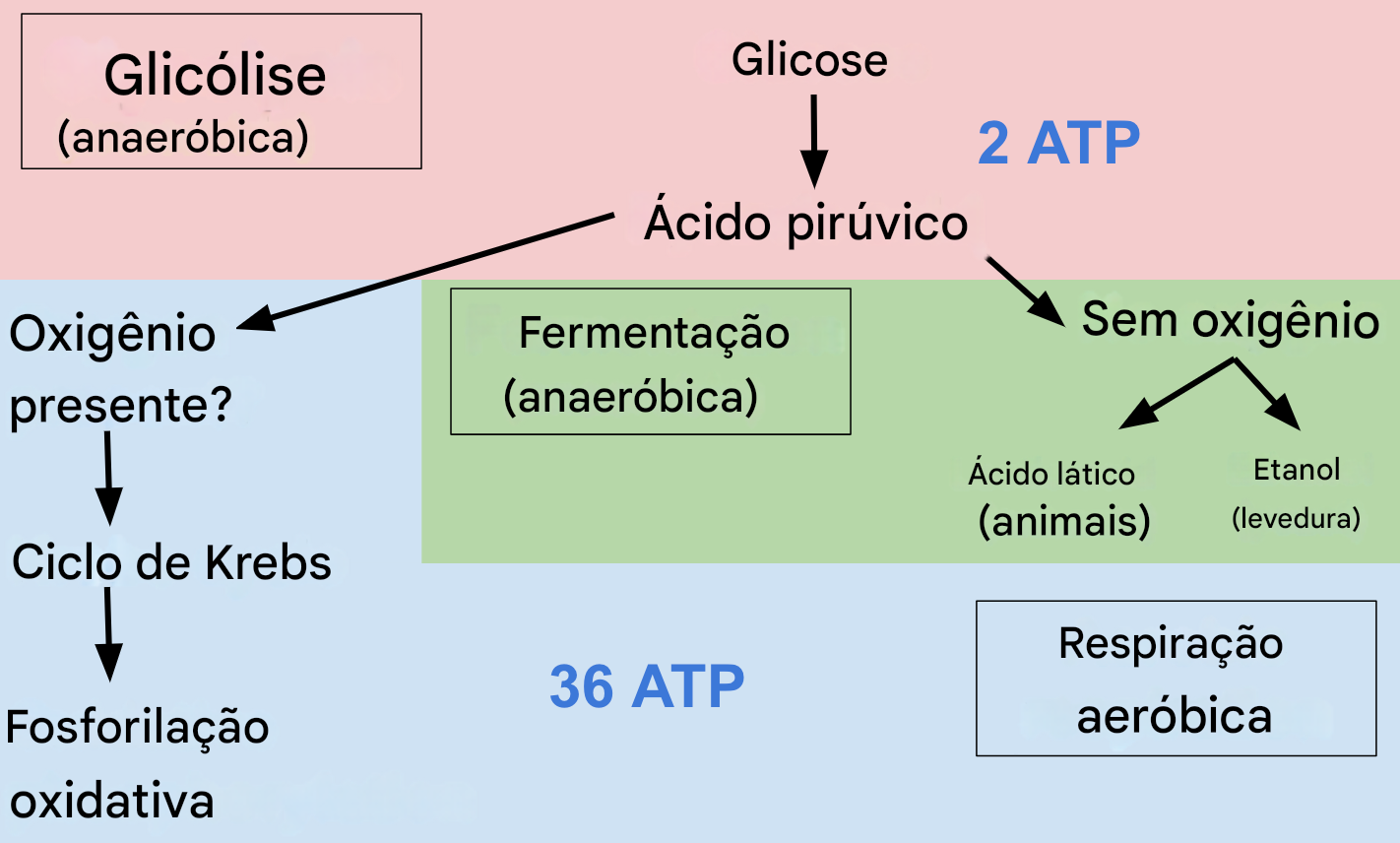
Reprogramação metabólica: glicólise aeróbica
O cérebro no TEA apresenta um padrão metabólico semelhante ao observado em células proliferativas:
aumento da glicólise aeróbica (efeito Warburg), com síntese de 2 ATPs
Ao contrário da glicólise anaeróbica que ocorre sem oxigênio, a partir da transformação de piruvato em lactato, a glicólise aeróbia, ocorre com oxigênio. O objetivo é mais que formar ATP, é formar intermediários para síntese de nucleotídeos, lipídeos e aminoácios (ver abaixo).
menor dependência da fosforilação oxidativa. Isso é problemático, pois a energia será rápida, mas insuficiente.
Consequências do efeito Warburg
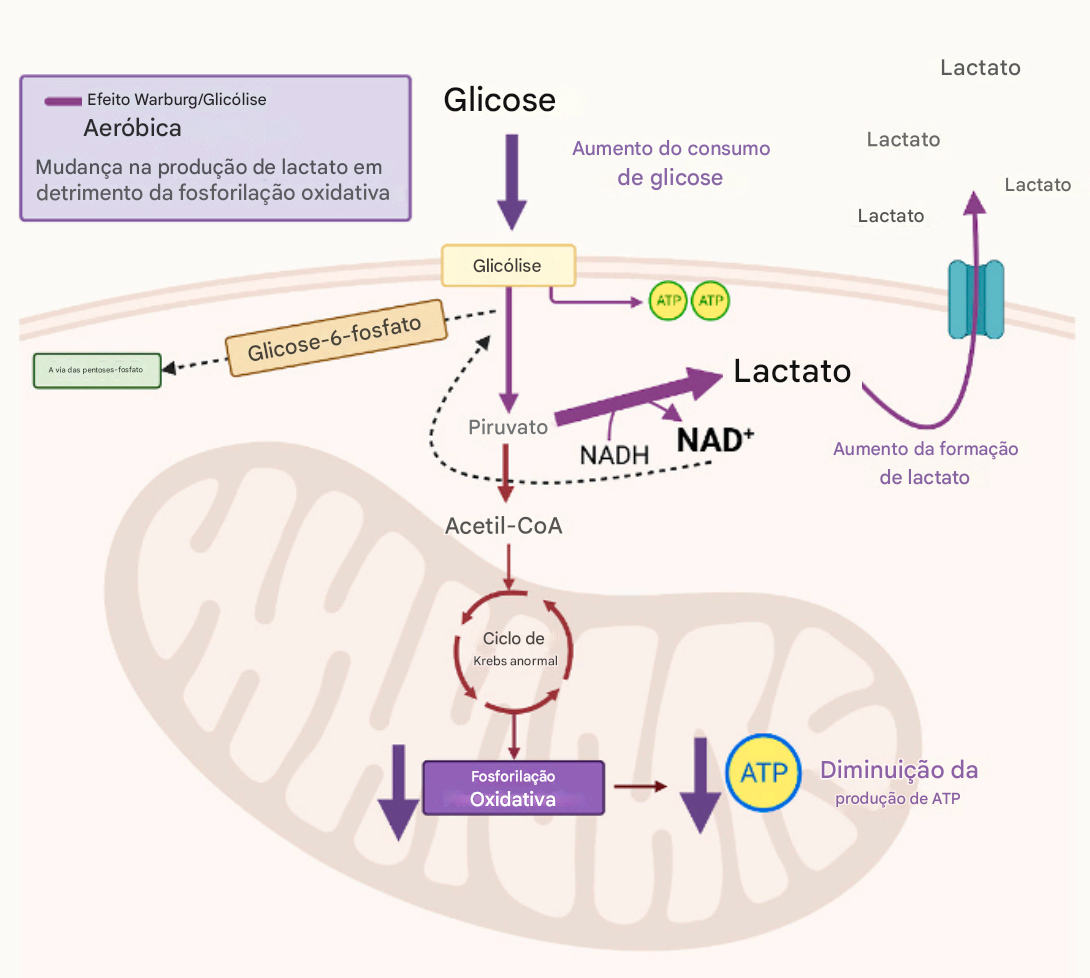
produção rápida de energia (2 ATP), para células em alta demanda
suporte à proliferação celular anormal
A proliferação celular não depende apenas de ATP, mas também de nucleotídeos (DNA, RNA), aminoácidos e lipídios de membrana
A glicólise anaeróbia fornece intermediários metabólicos (glicose-6-fosfato, piruvato, ribose-5-fosfato, NADPH) e matéria prima para biossíntese de nucleotídeos, membranas, etc
possível impacto na maturação neuronal
Neurônios imaturos são mais dependentes de glicólise aeróbia
Neurônios maduros são mais dependentes de fosforilação oxidativa mitocondrial
Essa mudança metabólica é induzida pela ativação da via WNT/β-catenina. A glicólise aeróbica elevada durante fases tardias do desenvolvimento pode manter neurônios em estado metabólico imaturo, interferindo na transição para um fenótipo neuronal maduro e funcionalmente estável.
Integração dos mecanismos
O modelo proposto sugere:
Disfunção circadiana
→ desregulação da via WNT/β-catenina
→ reprogramação metabólica cerebral
→ alterações no neurodesenvolvimento e comportamento
Trata-se de um sistema interdependente. O estudo redefine o TEA como resultado da interação entre tempo biológico, sinalização celular e metabolismo. A desorganização do ritmo circadiano não é apenas um sintoma, mas um possível fator causal que altera profundamente o desenvolvimento cerebral.
Implicações clínicas e científicas
O TEA pode ser parcialmente compreendido como um distúrbio cronobiológico-metabólico
Intervenções futuras podem focar em:
regulação do sono e luz
modulação metabólica
alvos moleculares da via WNT
É importante lembrar que tudo já começa no ambiente intra-uterino (na barriga da mãe). O metabolismo fetal é altamente dependente do ambiente materno:
glicemia e níveis de insulina modulam indiretamente sinalização WNT, estado redox, preferência metabólica celular.
inflamação (IL-6, TNF-α), muda atividade de vias de crescimento e sinalização do desenvolvimento neural. Ambiente inflamatório desorganiza timing de diferenciação, equilíbrio proliferação vs maturação.
disponibilidade de nutrientes, como B6, B9, B12 para adequada programação fetal, metilação de DNA, acetilação de histonas. Esses sistemas regulam a sensibilidade a WNT, expressão dos genes do relógio, metabolismo celular.
Assim, gestante precisa dormir, comer bem, receber suplementação adequada, cuidar da saúde. Após o nascimento continuam as intervenções neurofuncionais, comportamentais e de estilo de vida (incluindo higiene do sono) para apoiar plasticidade cerebral, com impacto em redes neurais, linguagem, cognição e autorregulação.
O cérebro pós-natal é plástico. Sinapses são remodeláveis, redes neurais são ajustáveis por experiência, função pode ser otimizada mesmo com organização atípica inicial.
O sono estabiliza ritmos circadianos, melhora consolidação sináptica. Sono adequado + nutrição + atividade física + terapias não revertem o TEA, mas propiciam ganhos. O que queremos é a reorganização funcional de circuitos neurais por plasticidade.